技术交流

扫描二维码
或添加“GeneGroup003”
获取更多更新资讯
商城订购

扫描二维码
或添加“基因商城(GeneMart)”
手机下单,快人一步
售后服务

扫描二维码
或添加“GeneGroup005”
获取更快速售后支持
泛基因组经典文章回顾——PacBio SMRT助力大豆泛基因组研究,打开遗传变异分析新思路

“工欲善其事,必先利其器”
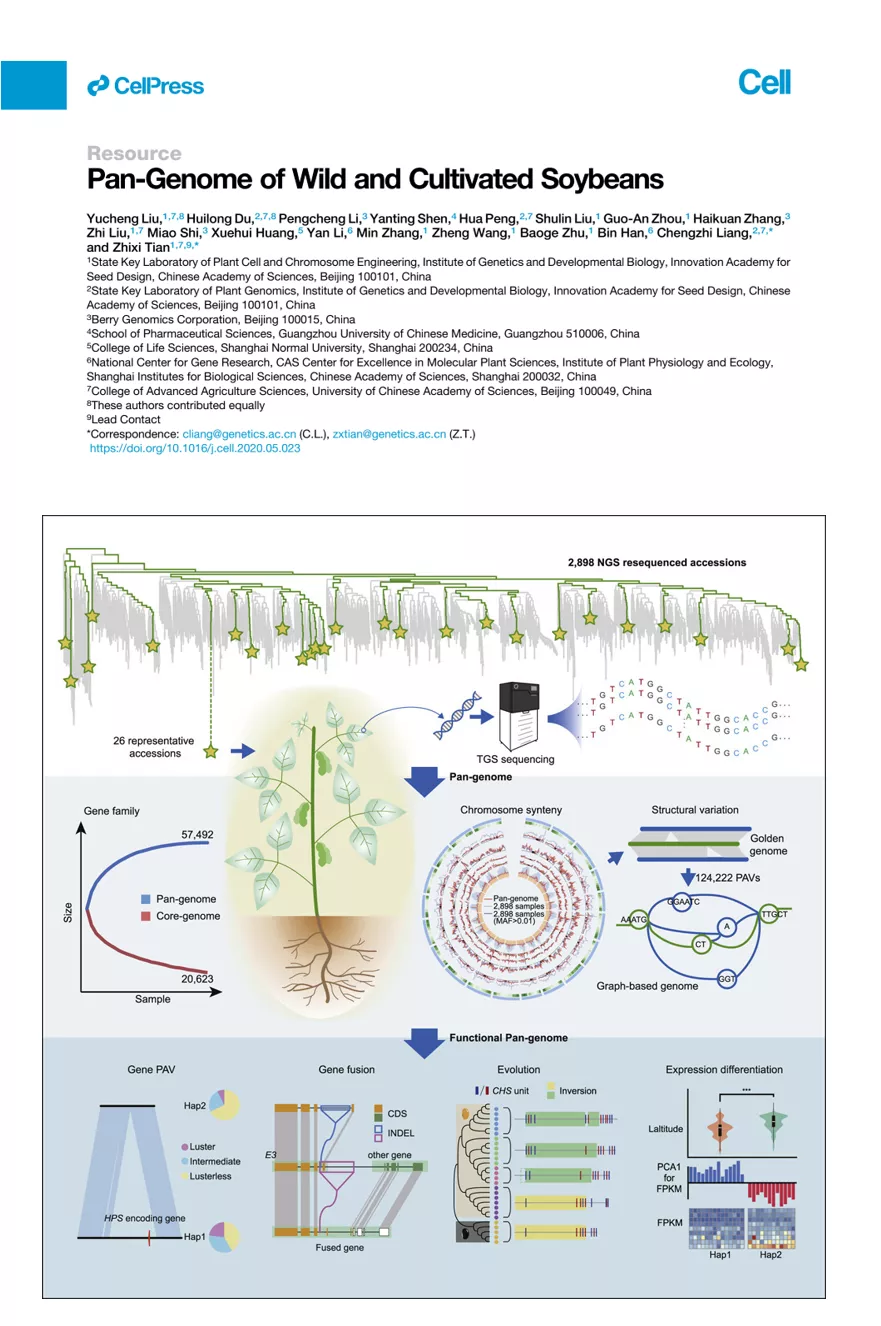
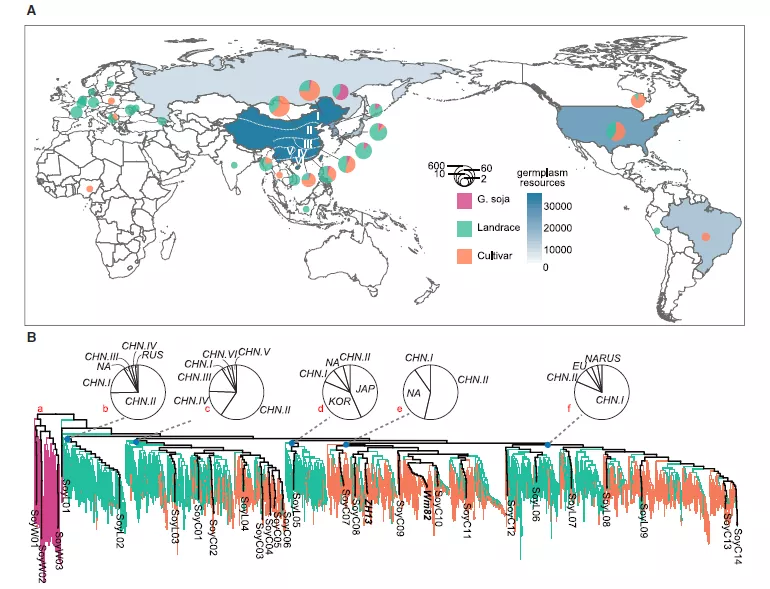
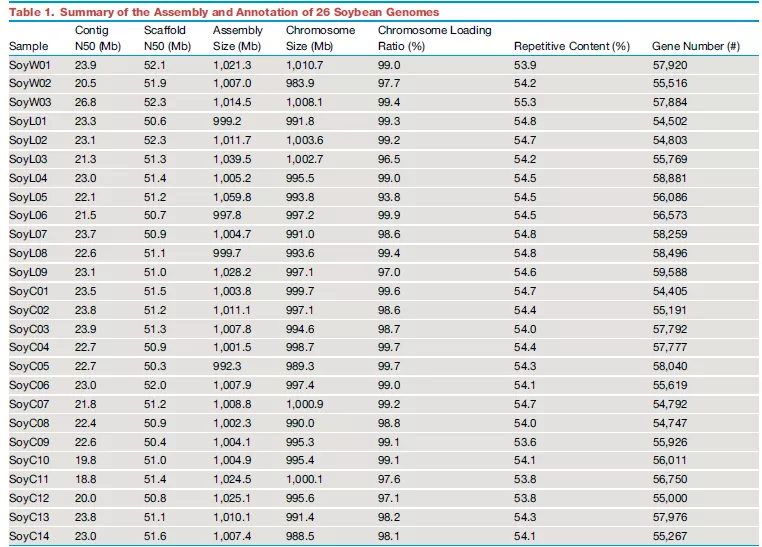
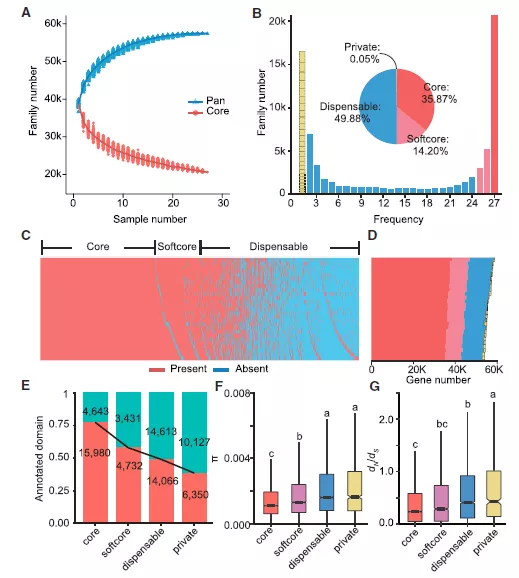
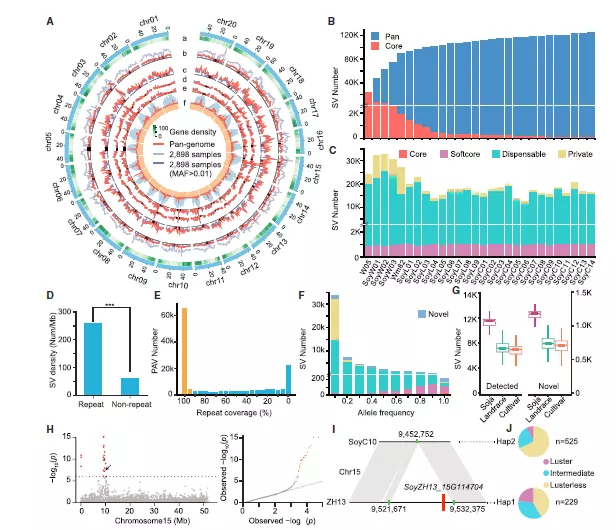
PacBio 三代测序技术可以获得长读长且准确的序列,可鉴定出短读长测序技术无法识别的长片段遗传变异信息,成为泛基因组组装的一大利器。 大豆是国家重要的粮食经济作物,为人类提供了主要的油料和蛋白资源。目前,被广泛应用的参考基因组有三种:Williams 82、Zhonghuang 13、W05。通过对三种参考基因组的比较,发现大量结构变异,比如获得与缺失变异(PAVs,presence/absence variants)和拷贝数变异(CNVs,copy number variants),导致单一或少数基因组不能完全代表一个物种所有的遗传变异。因此需要构建能够广泛代表不同大豆品种的泛基因组。 Cell文章—植物泛基因组 中国科学院遗传与发育生物学研究所田志喜研究团队长期从事大豆功能基因组研究,2020年7月9日在Cell上发表了题为Pan-Genome of Wild and Cultivated Soybeans的文章,通过PacBio长读长测序技术对26份具有代表性的野生和栽培大豆的基因组进行从头组装,构建了一个高质量的大豆图形结构基因组,并进行泛基因组分析,发现了大量通过短读长测序技术没有发现的遗传变异,有助于识别与农艺性状相关的候选基因。 ● 样本选择策略 为了构建能代表大豆全部遗传多样性的泛基因组,作者对2898份大豆样本进行illumina 13×测序(2898份大豆样本中包括103份野生大豆、1048份农家种和1747份栽培品种,并在全球范围内收集,代表了大豆地域分布),与参考基因组Gmax_ZH13比对,鉴定出31,870,983个SNPs,通过SNP构建系统发育树,结合地域分布进行分析,挑选出最能代表2898份材料,以及对生产和育种有较大贡献的26份大豆样本,包括了3份野生大豆,9份农家种和14份栽培品种,进行泛基因组的构建。 图1.2898份重测序大豆材料的地理分布及系统发育分析 A.2898个品种的地理分布。各地区收集的材料数量用饼图的大小表示,各地区野生大豆(紫色表示)、农家种(绿色表示)、栽培品种(橙色表示)的比例在饼中显示。每个国家或地区的大豆种质资源总数以蓝色深度表示。 B.根据全基因组SNPs推断得出的系统发育树。不同颜色的线条代表不同的种类,野生大豆(紫色)、农家种(绿色)、栽培品种(橙色) ● 测序策略 对挑选出来的26个大豆样本进行PacBio 单分子实时(SMRT,single molecule real-time)测序,平均深度为96X;结合平均深度为277X的Bionano 光学图谱数据和染色体构象捕获(Hi-C)以及illumina 测序数据,进行了全基因组组装和注释。所得到的contig N50平均长度达22.6M,scaffold N50平均长度达51.2M,组装的基因组大小平均长度达1011.6M。 表1.26个大豆基因组的组装与注释综述 ● 泛基因组分析 对26份大豆样本和zhonghuang13参考基因组进行泛基因组分析,从图2(A)中可以看出随着基因组数目的增加,泛基因组中的基因数量也随之增加,当基因组数目达到25个时,基因数目增加到平台期。因此由这27个基因组所构建的泛基因组可以覆盖大豆所有的基因。根据基因在27份泛基因组中存在的频率定义出Core Genes(存在27份样本中)、Softcore Gene(存在25-26份样本中)、Dispensable Genes(存在2-24个样本中),Private Gene(仅存在单个样本中),结果比例如下图。虽然Dispensable Gene和Private Gene的个数多,但是在单个样品中比例不高。可以看出Core Gene和Softcore Gene在50%左右,Dispensable Gene为50%左右(如图2B-2D)。Core gene有更高的domain比例,同时π和dn/ds也比较低,说明Core Gene更保守(如图2E-2F)。 图2.27份大豆样本的泛基因组和核心基因分析 A.随着大豆样本数量的增加基因家系中泛基因组和核心基因的变化;B.泛基因组和个体基因组的组成;C.27个大豆基因组中泛基因组的存在与缺失信息;D.个体基因组中各成分的基因数;E.Core、softcore、dispensable、private 基因中具有InterPro domains的基因比例;F.Core、softcore、 dispensable、private 基因中核苷酸多样性(π)和dN/dS的比例。 ● 构建图形结构基因组并分析结构变异 作者认为如果构建多个泛基因组而不进行下一步的整合将无法打破不同个体之间遗传多样性的瓶颈。因此,他们首次尝试基于图形结构将不同的泛大豆基因组整合成为一条完整的泛基因组,打破了传统线性基因组的储存形式。 作者将29个大豆样本(26个denovo组装和Williams 82、Zhonghuang 13、W05)的基因组整合成一条图形结构基因组(graph-based genome),将鉴定到的776,399个SVs进行合并成124,222个非冗余SVs。与核心基因组的定义思路相似,按照SVs在不同样本中出现的频率定义为Core 、softcore、Dispensable,private gene。研究中发现Wm82有较高比例的private SVs,作者认为这种情况可能是用二代测序技术组装的原因。同时发现SVs更倾向于存在DNA重复的区域。比以往发现了更多的PAVs,78.5% 的PAVs都来自DNA重复区域。作者将2898个大豆重测序数据比对到graph-genome上,共鉴定到55,402个SVs。从2898份材料中鉴定到3584个新的SVs,野生大豆中鉴定到的SVs要明显多于地方种和栽培种。基于图形结构基因组分型的SVs对种子光泽进行的全基因组关联研究(GWAS),确定了15号染色体上的一个重要信号,其中一个10 kb的PAV导致了一个疏水蛋白(HPS)编码基因的存在和缺失,进一步分析含有和不含有这10 kb序列的大豆种子分别具有较高比例的光泽和无光泽,这表明PAV可能是控制大豆种子光泽变化的因果遗传变异之一。 图3.29份大豆基因组和2898份重测序样本的遗传变异 A.29份大豆基因组和2898份重测序样本的遗传变异分布;B.随着样本数量增加,样本中的变异数量的变化。(以非冗余策略进行合并);C.每个样本中不同类别的变异的数量;D.重复和非重复区域中结构变异的密度;E.DNA重复区域的数量与结构变异数量的关系;F.等位基因的频率与结构变异数量的关系。结构变异是通过将短读长测序比对到图形结构基因组上检测出的;G.从野生大豆,农家种和栽培品种中发现的新SVs的数量;H.基于图形结构基因组进行GWAS测序;I.一个编码疏水蛋白(HSP)的10kbPAV;J.10 kbPAV的两个单倍型种子光泽变异的比较. ● PacBio 强势助力泛基因组研究 HiFi 测序是PacBio测序平台推出的兼顾长读长和高准确度的测序技术。作为PacBio最新的数据类型,既兼顾读长(20kb的长度)又具有高准确度(>99.9%准确率)的HiFi reads,不仅可以改善变异检测,减少组装时间,还能够识别复杂的基因组区域的细微差别,有助于增加基因组组装的连续性,准确性和完整性,可以实现多倍体基因分型。在泛基因组研究中可以提供高质量的参考基因组,同时可以有效节约生信分析的时间,大大加速大型泛基因组研究的效率。 从待研究物种群体中选择具有足够遗传多样性的代表来捕捉大部分变异,通常20-30品系可以完成该工作。然后对这20-30个品系进行全基因组测序以及全长转录组测序,从基因组层面和转录组层面打造高质量参考基因组。接着就可以在剩余的物种内进行低覆盖率的重测序,以进一步分类每个品系并捕获全部的变异。 参考文献:Liu Y , Du H , Li P , et al. Pan-Genome of Wild and Cultivated Soybeans[J]. Cell, 2020, 182(1). 更多资讯请点击: 为了更好的向您推送PacBio SMRT测序技术的最新进展,基因有限公司开辟了专门的企业微信账号,实时解读最新PacBio研究进展。 长按并识别二维码并添加联系人,您将了解到有关高准确度长读长测序技术的最新进展,以及我们即将举办的线上线下活动。 基因有限公司作为PacBio公司在中国区的独家代理商,自2011年以来将PacBio第三代单分子实时测序技术引入国内,一直为国内用户提供专业的三代测序系统的安装培训,技术支持,应用培训与售后维护工作,赢得客户的一致好评与信任。基因有限公司将一如既往的支持越来越多的PacBio用户。