
技术交流

扫描二维码
或添加“GeneGroup003”
获取更多更新资讯
商城订购

扫描二维码
或添加“基因商城(GeneMart)”
手机下单,快人一步
售后服务

扫描二维码
或添加“GeneGroup005”
获取更快速售后支持
PacBio HiFi vs. ONT,谁能更好地解析环境真核微生物中的rRNA操纵子?

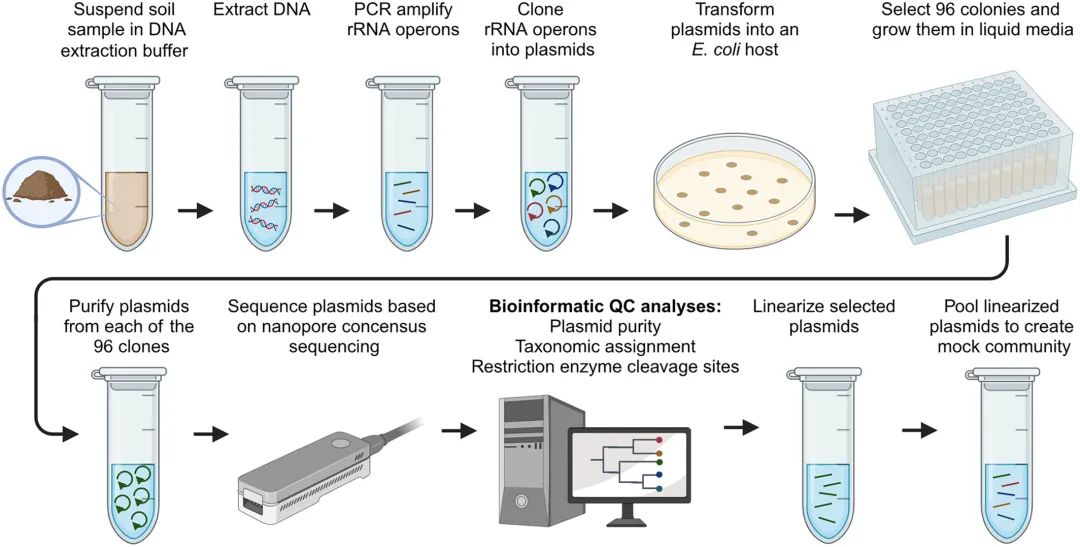
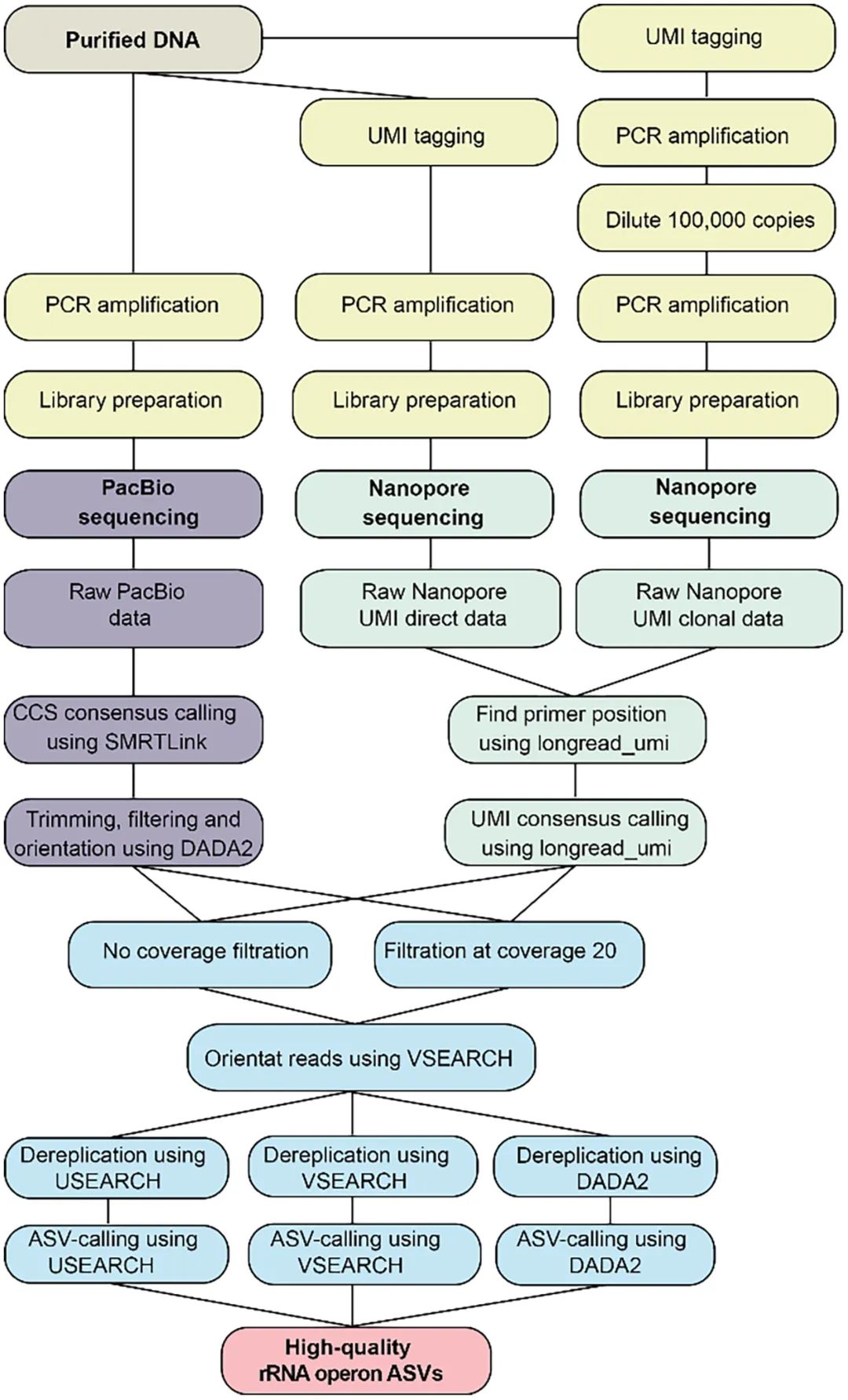
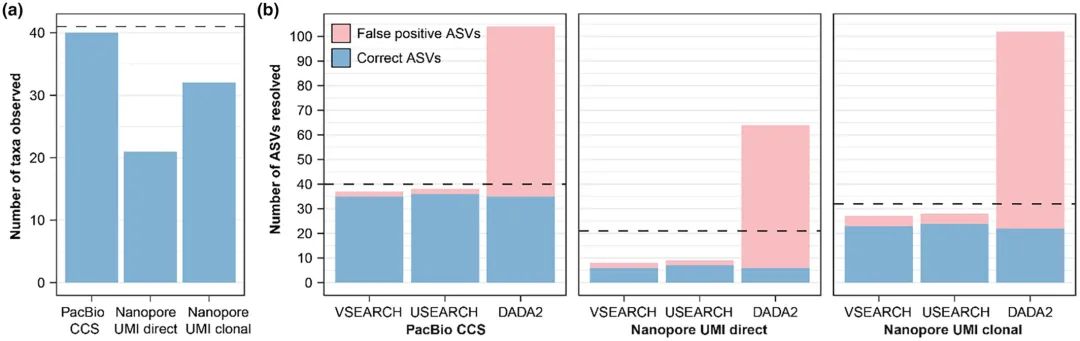
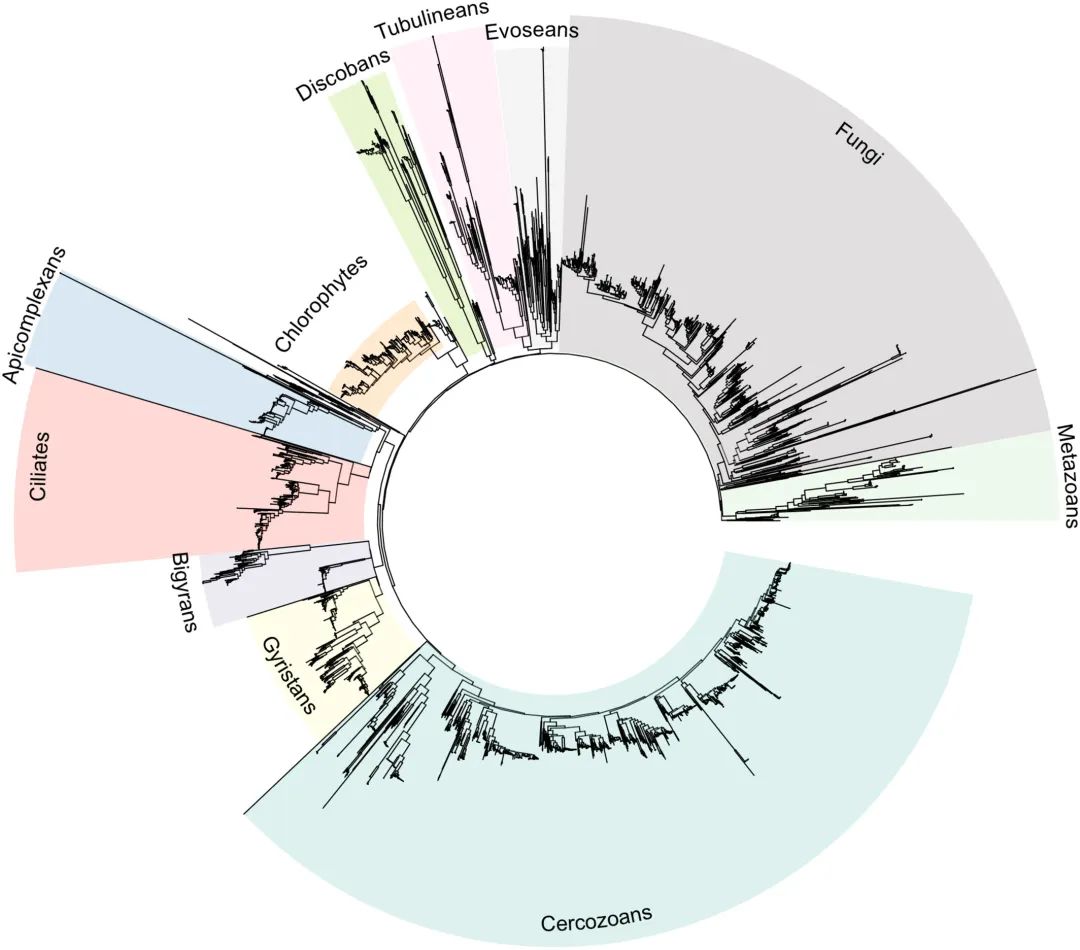
真核微生物的分类研究 真核微生物的分类研究通常基于短读长宏条形码测序(原生生物18S rRNA的V4/V9区域或真菌的ITS1/ITS2区域),虽然其已经彻底改变了研究真核微生物群落的方式,但短读长的分类学和系统发育识别能力有限,现在更加需要基于全长18S rRNA组成的参考数据库。此外,现在仍缺乏全面的18S rRNA参考数据库。这限制了对不同测序技术和生物信息学处理性能的评估。 随着长读长测序的使用越来越多,评估各种测序方法和生物信息学工具分析扩增子序列变异(ASV)的准确性是至关重要的,以确保最终序列代表真正的生物变异。为了解决这一挑战,奥尔堡大学化学与生物科学系微生物群落中心的研究人员创建了一个高度多样化的真核微生物模拟群落,用来评估三种长读长测序策略(PacBio CCS循环一致性测序和两种使用独特分子标识(UMI)的Nanopore方法)和三种分析ASV的工具(USEARCH, VSEARCH和DADA2)的性能。相关成果已发布“Benchmarking long-read sequencing strategies for obtaining ASV-resolved rRNA operons from environmental microeukaryotes”。 长读长测序技术现状 长读长测序技术 PacBio测序使用环形一致性测序(CCS)对单个环化的DNA分子进行多次测序,产生精确的一致性序列(单分子准确度>99.9%,一致性准确率>99.999%)。由于准确度高、读长长,该方法已广泛应用于人体肠道研究、环境研究和医学研究等多样性领域。 Nanopore测序则是基于DNA分子通过膜嵌入的孔时的电流变化直接读取(一次电流信号代表4-5个碱基),实时测序使得其可以提供快速的检测结果,多适用于流行病学研究等。而目前对于准确度上的补足则是采用引入唯一分子标识符(UMIs)来纠正原始Nanopore数据中较高的错误率。目的是区分扩增过程中产生的PCR克隆和原始样品中的独特分子。 生信工具 所有的测序方法都不可避免地产生有一定程度误差的序列。将这些错误与真正的生物变异区分开来是至关重要的。这种区分是使用生物信息学工具实现的,特别是扩增子序列变异(ASV)解析工具(USEARCH, VSEARCH和DADA2)。目前已经为PacBio CCS数据开发了DADA2流程。但目前还没有用于Nanopore数据的DADA2流程,研究人员表示非常担忧由高错误率数据组成而创建出错误的模型。 结果与讨论 长读长测序技术分析真核微生物模拟群落的准确性 研究人员通过将土壤真核微生物中PCR扩增的rRNA操纵子克隆到质粒中,构建了一个合成的高度多样化真核微生物模拟群落,来比较不同长读长技术PacBio CCS和Nanopore UMI*获得的高质量真核微生物rRNA操纵子的表现(图1)。 图 1. 构建真核微生物模拟群落的流程。 *Nanopore UMI创建了两种不同类型文库:Nanopore UMI直接文库和Nanopore UMI克隆文库(图2)。直接文库涉及在UMI标记后立即进行一轮扩增,而克隆文库涉及使用Nanopore UMI直接文库中定义数量的UMI标记扩增子作为模板,进行第二次克隆扩增。然而要注意克隆方法中的两步扩增可能引入嵌合体,这在理论上不应该发生在直接方法中。 图2. 用于PacBio和Nanopore数据的文库制备和生物信息学流程。为不同测序方法制备的测序文库以黄色显示。PacBio reads的独特处理以紫色表示。 经过过滤,研究人员分别获得了1,250,905个PacBio CCS reads,以及169,632和41,016个Nanopore UMI一致性序列。为了避免测序深度的带来的分析偏差,研究人员将所有数据集精简为相同数量的一致性reads(41,016个序列)。在相同的测序深度下,PacBio CCS对42个分类群中的40个模拟群落提供了最好的序列覆盖率。相比之下,Nanopore UMI方法仅为直接和克隆数据集分别提供了21和32个序列的覆盖*(图3a)。 *研究人员认为Nanopore UMI数据的低覆盖率与UMI标记效率不佳有关,很可能是由于标记引物上的40 bp的突出部分(overhangs)导致的。 图 3. 在模拟群落中识别 ASV。 不同生信工具解析真核微生物模拟群落ASV的准确性 解决ASV时要考虑的关键因素是如何从PCR和测序产物中辨别真正的生物变异。因此解析ASV需要无错误序列,研究人员检查了与模拟群落完全匹配的CCS和UMI一致性序列的百分比,发现一致性序列的准确性受到PacBio的CCS轮数和Nanopore的UMI- bin原始reads数的影响。在PacBio和Nanopore数据中,完美序列的百分比在覆盖率约为20×时达到平稳期,并且PacBio CCS数据的准确性(~35%完美reads)通常比Nanopore UMI直接(~20%完美reads)和克隆(~10%完美reads)数据更好(图4)。 图4. 每个CCS/UMI覆盖的完美一致性序列的百分比 USEARCH, VSEARCH和DADA2解析ASV的表现: 与之前的覆盖率分析一致,PacBio CCS数据中正确ASV数量最多,其中原始数据中发现的40个唯一参考序列中有36个可以使用USEARCH识别,同时只调用了两个假阳性ASV。VSEARCH的表现几乎和USEARCH一样好,只少检测到一个ASV。DADA2能够检测到35种ASV,同时引入69个假阳性ASV。 对于Nanopore UMI直接数据,ASV的数量惊人地低,USEARCH在原始reads覆盖的21个ASV中,最多只能识别出7个正确的ASV。尽管使用了UMIs,仍观察到少数ASV假阳性。VSEARCH产生了类似的结果,但遗漏了一个USEARCH检测到的正确ASV。 对于Nanopore UMI克隆数据,正确的ASV数量要高得多,使用USEARCH在原始数据覆盖的32个ASV中识别出24个正确的ASV。然而,假阳性ASV的数量要高得多,这反映了UMI一致序列的准确性较低。DADA2能够检测到与VSEARCH相同数量的正确ASV,但假阳性ASV也很多。UMI数据集中出现的调用差距可能归因于UMI标记不足。 综上,通常建议使用PacBio CCS数据基于USEARCH或VSEARCH来解析rRNA操纵子ASV。 对于PacBio CCS和Nanopore UMI克隆数据,研究人员建议minsize为3,而对于Nanopore UMI直接数据,由于UMI合并过程中固有的嵌合体过滤,可以minsize为2。 高质量的长读长数据在真实土壤样本的表现 接着,研究人员在丹麦阿斯科夫的真实农业土壤样本上复刻了上面描述的真核微生物群落的测序和分析。从该样本中,研究人员获得了335,947个PacBio CCS序列,31,693个NanoporeUMI直接序列和16,257个NanoporeUMI克隆一致序列。为保证一致性,所有数据集都被精简为16,257个序列,使用上面推荐设置完成ASV调用。然后合起来分析来自三种类型测序数据的ASV,共获得了5,123个独特的近全长rRNA操作子。 rRNA操纵子几乎包括了所有主要的真核生物(图5):多样性最高的是真菌和丝足虫(Cercozoans),其次是纤毛虫(Ciliates)、顶复动物(Apicomplexans)、旋毛虫(Gyristans)、绿藻(Chlorophytes)和变形虫(Amoebozoans)。该研究是首批分析微真核生物的ASV解析rRNA操作子的研究之一,进一步提取其18S rRNA基因,发现分别有59.5%、40.9%和21.1%的序列代表潜在的新种(<99%一致性)、属(<97%一致性)和科(<93%一致性)。这凸显了在土壤中仍有待检测和研究的大量未被发现的真核微生物多样性。 图5. 丹麦阿斯科夫土壤中的真核微生物的多样性。 讨论与建议 在这项研究中,研究人员开发了一种构建合成真核微生物模拟群落的方法,然后基于PacBio CCS或Nanopore UMI一致性reads,为如何从真核微生物中获得高精度的ASV解析rRNA操纵子提供建议。并应用于一个复杂的农业样本完成5,123个独特的ASV解析rRNA操纵子的鉴定,为研究土壤中隐藏的真核微生物多样性提供了新思路。 基于这项研究的发现,引入UMI的方法似乎是研究更复杂样本的一种有前途的方法。然而,考虑到使用UMI引物扩增相关的挑战(引物的长度和严格的DNA质量要求),PacBio方法更加是首选,其需要更少的手工操作,显示出更好的稳健性和可重复性,没有UMI标记引入的随机偏差。 Nanopore UMI测序的挑战 Nanopore测序的高错误率是限制ASV调用的关键因素。为了提高Nanopore测序的准确性,研究人员使用了UMIs来减少测序误差。然而,UMI标记引入的随机偏差和长UMI引物PCR带来的困难会导致结果不一致。如UMI太少会导致分类群多样性低,而太多则会导致覆盖不足。即使经过调整,NanoporeUMI克隆方法靶向所需数量的分子仍然具有挑战性。因此,使用UMIs是在获得无嵌合体数据和可能丢失的分类群之间的折衷。 本文使用的PacBio sequel II与Nanopore MinION测序的比较分析 研究人员评估了从测序成本到数据处理时间和数据质量的各种参数(表2)。 在文库制备时间:PacBio文库制备更快,大约需要半天,而使用UMIs制备Nanopore文库大约需要1天。 样本质量要求:Nanopore测序和PacBio测序都需要高质量的DNA。然而,Nanopore测序有更严格的要求。因为DNA提取的污染物会破坏Nanopore,从而降低测序能力。即使是少量的杂质也会显著影响关键的UMI标记反应。 均聚物长度估计的误差:Nanopore数据中尤为明显。 数据处理:处理原始Nanopore reads需要大量的计算时间(在高性能计算机上为100周),在此期间会生成大量临时文件并运行进程;由于PacBio Sequel IIe和Revio都直接提供处理过的CCS,因此大大减少了生物信息学处理(最近推出的PacBio Revio显著提高了测序输出和准确性,允许以与Sequel II相同的成本从一个cell获得三倍以上的数据)。 以上这些优势,加上每个一致性序列的成本低得多,使PacBio平台成为当前的首选方法。 表1. PacBio和Nanopore技术在环境真核微生物rRNA操纵子高通量测序中的比较 参考文献:Overgaard, C. K., Jamy, M., Radutoiu, S., Burki, F., & Dueholm, M. K. D. (2024). Benchmarking long-read sequencing strategies for obtaining ASV-resolved rRNA operons from environmental microeukaryotes. Molecular Ecology Resources, 24, e13991. https://doi.org/10.1111/1755-0998.13991 关于我们 基因有限公司作为PacBio公司的中国区合作伙伴,自2011年以来将PacBio第三代单分子实时测序技术引入国内,一直为国内用户提供专业的三代测序系统的安装培训,技术支持,应用培训与售后维护工作,赢得客户的一致好评与信任。基因有限公司将一如既往的支持越来越多的PacBio用户。





关于基因 基因有限公司成立于1992年,是一家提供生命科学科研仪器、试剂耗材和技术服务的综合服务商。基于“Gene Brightens Every Life • BioTech Connects the World”——“基因燃亮生命 • 生物技术连接世界”的愿景,专注于生命科学领域前沿技术的引进和推广,致力于推动该领域国内科研机构硬件水平及实验方案的革新与升级。同时,公司也一直致力于自主品牌的科研设备的研发与生产,拥有一系列通用性强、互补性高的自主品牌产品。 基因的服务网络遍及全国各地十多个大中城市,拥有包括仪器销售,试剂销售,市场与技术支持,维修,客服,物流等多个部门组成的完整服务体系。 我们希望通过不懈努力,为您的成功铺路搭桥,也为中国的生命科学事业赶超世界先进水平尽一己之力。欲了解更多信息,请访问www.genecompany.cn。









