技术交流

扫描二维码
或添加“GeneGroup003”
获取更多更新资讯
商城订购

扫描二维码
或添加“基因商城(GeneMart)”
手机下单,快人一步
售后服务

扫描二维码
或添加“GeneGroup005”
获取更快速售后支持
PacBio HiFi 测序助力首个高精度中国人泛基因组数据发表

在过去的二十年中,人类参考基因组序列已成为遗传和生物医学研究与应用的基础。然而,有一个广泛的共识,即现有的单一的人类参考基因组序列无法表征全球人群的基因组多样性。
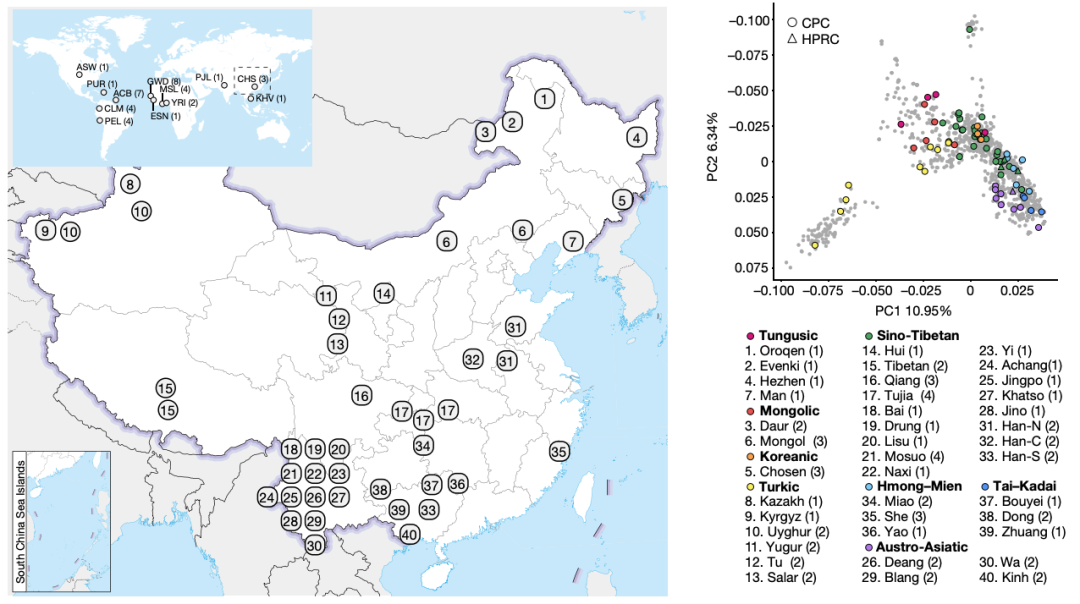
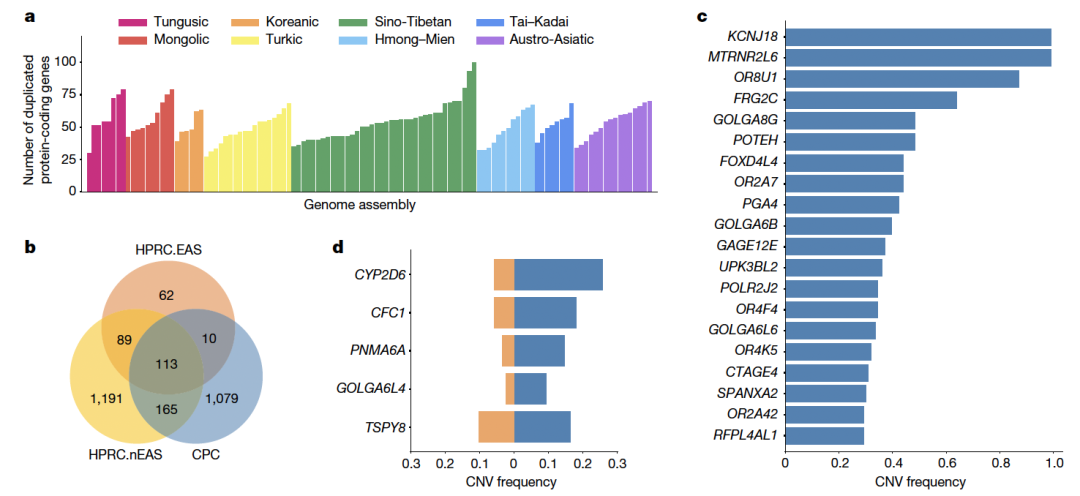
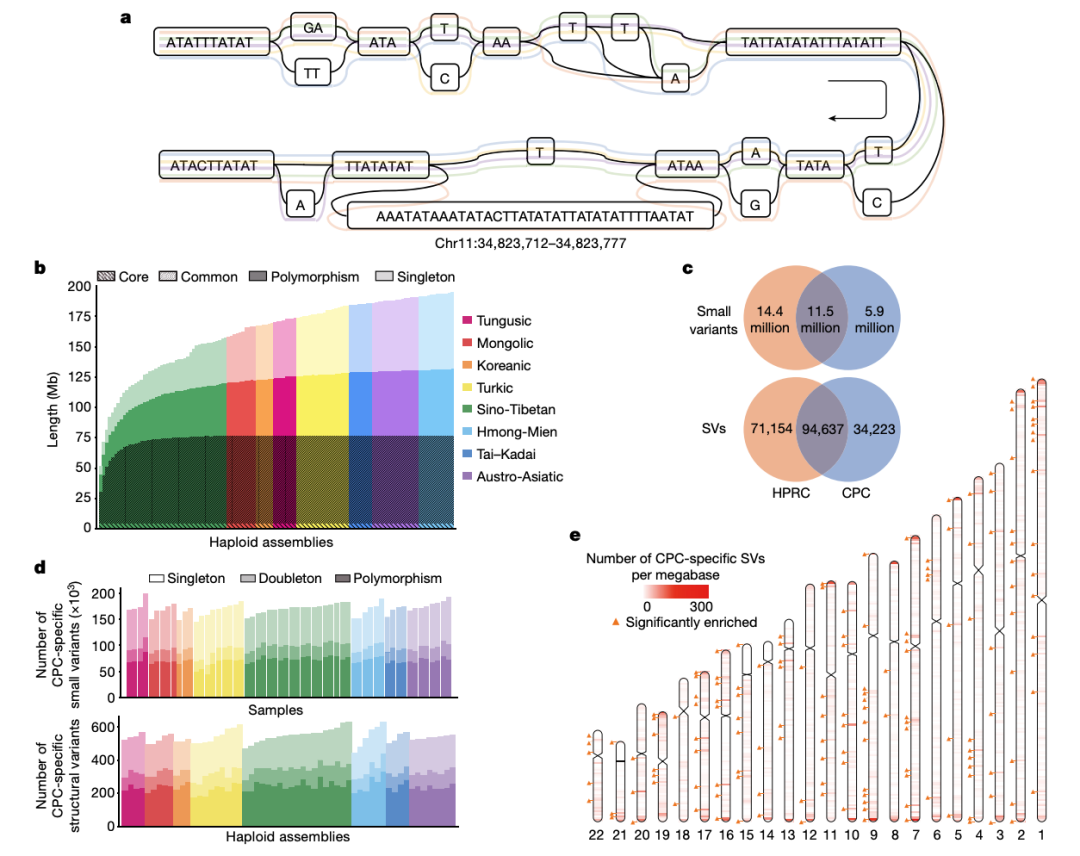
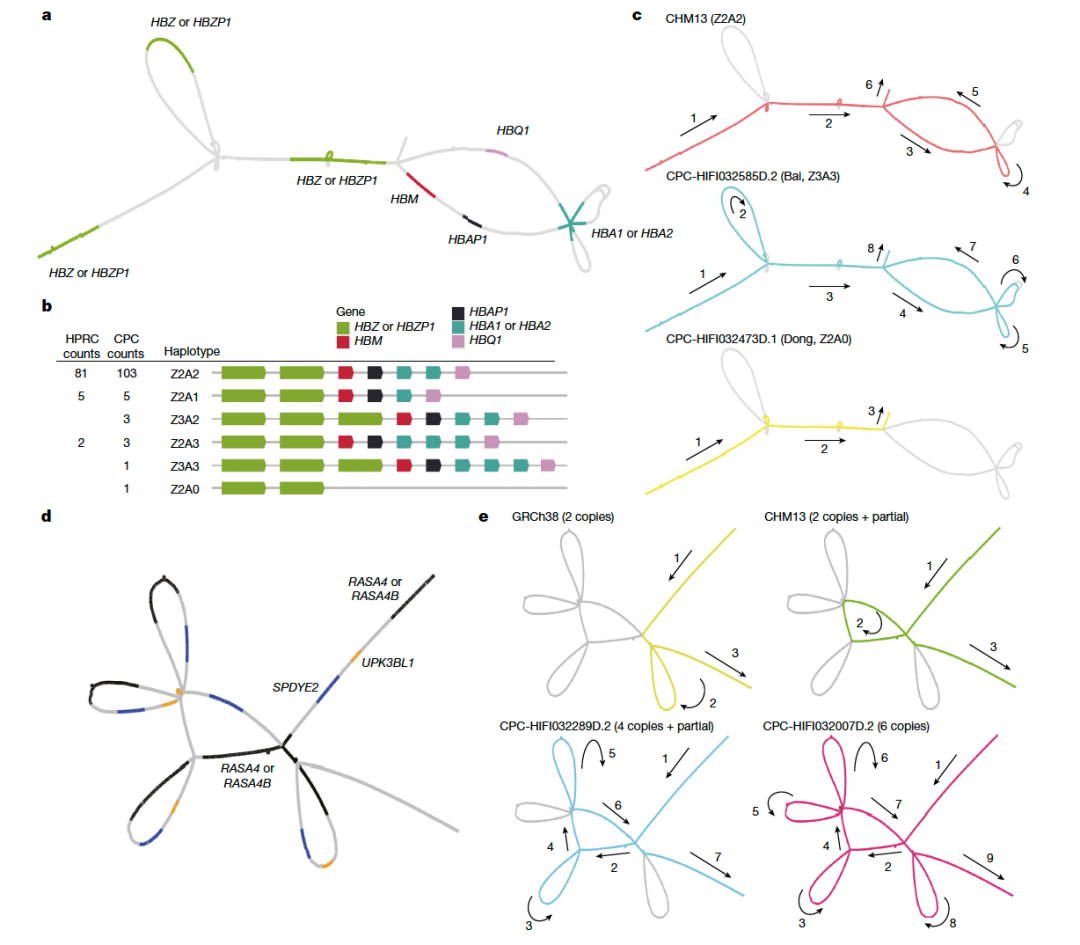
随着长读长测序技术和生信方法的进步,现在可以通过泛基因组来构建和捕获大量不同基因组中遗漏的变异。人类泛基因组参考联盟(HPRC)最近基于全球人群的47个样本构建了人类泛基因组参考草图,但东亚人群样本的代表性不足(n = 4)【1】。特别是HPRC文献中仅包含三个南方汉族(CHS)样本,数量太少,无法代表族群的基因组多样性。为了构建高质量高精度的中国人群泛基因组参考图谱,复旦大学徐书华教授、西安交通大学叶凯教授联合国内26家单位发起了中国人群泛基因组联盟(Chinese Pangenome Consortium, CPC)。 CPC一期核心样本地理分布及语系、族群、遗传聚类关系 为了能够更快速、准确地构建高质量的中国人群泛基因组数据,CPC主要使用了PacBio HiFi测序技术来对这36个中国不同民族的58个核心样本进行了长读长测序,并通过从头组装构建了116个高质量的人类基因组单倍型参考序列。平均HiFi测序深度为30.65 X,平均Contig N50长度为35.63M。 与目前最新也是最常被使用的人类标准参考基因组序列GRCh38 相比,CPC的数据增加了 1.9 亿个碱基对的常染色质多态性序列和至少 1367 个蛋白质编码基因的复制事件。一共发现了1590 万个小变异 (SNV + small Indel) 和 7.8 万个结构变异,其中 580 万个小变异 (SNV + small Indel) 和 3.4 万个结构变异并未在其它研究(包括HPRC)中被报道过。并发现通用参考基因组上缺失的参考序列富集了适应性演化和起源于远古人类的遗传变异,并且与角质化、紫外线辐射应激、DNA 修复、免疫反应以及寿命等表型或功能相关。这项研究也显示了,建立我国自己的人群泛基因组图谱十分必要。在重构人类演化历程、挽回复杂疾病研究时“丢失的遗传率”等研究和应用中,该图谱具有巨大的潜在价值。 CPC核心组装的一些关键发现还包括: 发现了新的变异,特别是在遗传差异较大的少数民族中,例如α-球蛋白基因簇中新发现的20kb与10kb缺失,这将为进一步研究中国人群贫血症的遗传机理和致病机制提供新的线索。 研究人员在CPC参考图谱中发现了相当大比例的古人来源基因序列,平均每个族群和每个样本中分别有约15 Mb和约9.5 Mb的古人来源新序列。这可能是前期开展大量研究却未在现代人基因组中发现的古人基因渗入序列,或将为东亚现代人基因组中的古人基因渗入研究乃至整个古DNA领域提供新的信息资料和线索。 CPC泛基因组图谱中新发现了1079个基因拷贝数变异,以及包含药物代谢基因CYP2D6等在内的在中国人群中富集而在其他世界人群中出现频率较低的若干基因拷贝数变异;新鉴定出富集在中心粒、端粒等染色体复杂区域的3.4万个结构变异,其中半数以上仅在单个或两个样本中出现。 从CPC组件中识别到的CNV事件 得知文章发表,PacBio第一时间对徐书华教授和叶凯教授进行了采访,谈到这篇文章,两位老师表示: 徐书华 教授 复旦大学 CPC泛基因组参考文献无疑提供了对亚洲人群,尤其是中国血统人群基因组变异的更全面理解。例如,与人类泛基因组参考联盟 (HPRC) 最近发布的泛基因组参考相比,大约 18.4% 的小变异 (SNV + small Indel) 和 17.1% 的 SV 是特定于 CPC 组件的。这些发现对于未来基因组学研究,特别是人类进化史和医学遗传学领域具有重要意义。通过提供对亚洲人群遗传多样性的更全面理解,CPC数据可以帮助研究人员确定新的药物开发和个性化医学的靶点。此外,新序列和结构变异的发现可以帮助研究人员更好地理解复杂疾病和性状的遗传基础。 图形化泛基因组示例及CPC特有复杂变异分布 叶凯 教授 西安交通大学 相比传统线性基因组,泛基因组以图基因组方式刻画多个种族的遗传多样性,其内建先验变异信息为疾病研究提供潜在功能变异集合,解决领域面临的疾病研究丢失遗传性(missing inheribility)瓶颈。构建泛基因组通常有基于组装和变异比对两个流派,项目组开发了复杂结构变异检测软件SVision等自主知识产权计算方法,为本项目的成果提供了关键技术支撑。为了解析基因组中高重复高复杂区域,在数据收集方面,我们采用长读长、高精度的HiFi数据作为主体,采用组装和比对策略并重的方式,成功解析了人类基因组着丝粒序列和HOR结构。 相较于传统的短读长测序,PacBio HiFi 测序具有以下优势: 1. 卓越的测序读长:HiFi测序的读长是短读长测序的100倍,可以更好地解决基因组中的重复序列和结构变异。 2. 平均Q33准确率:HiFi测序具有更高的准确性,可以减少测序错误和假阳性结果。 3. 更好地解决基因组上的复杂区域:HiFi测序可以更好地解决复杂基因组,例如人类基因组中的长片段重复序列和结构变异。 4. 解析基因组单倍型:HiFi测序可以对单个样本进行单倍型解析,而无需进行家系测序。可以更好帮助科学家理解遗传多样性。这对于研究疾病和性状的遗传基础非常重要。 5. 直接检测DNA碱基修饰:HiFi测序在检测DNA序列的同时可以通过酶动力学信息获得DNA碱基修饰信息。可同时获得基因组和表观遗传学信息而无需额外的重亚硫酸盐处理步骤。 总的来说,HiFi序技术的优势使之成为泛基因组研究的首选方法,可以更好地解决复杂基因组和解析遗传多样性。 参考文献: Liao, WW., Asri, M., Ebler, J. et al. A draft human pangenome reference. Nature 617, 312–324 (2023). https://doi.org/10.1038/s41586-023-05896-x Gao, Y., Yang, X., Chen, H. et al. A pangenome reference of 36 Chinese populations. Nature (2023). https://doi.org/10.1038/s41586-023-06173-7 基因有限公司作为PacBio公司中国区代理商,自2011年以来将PacBio第三代单分子实时测序技术引入国内,一直为国内用户提供专业的三代测序系统的安装培训,技术支持,应用培训与售后维护工作,赢得客户的一致好评与信任。基因有限公司将一如既往的支持越来越多的PacBio用户。