
技术交流

扫描二维码
或添加“GeneGroup003”
获取更多更新资讯
商城订购

扫描二维码
或添加“基因商城(GeneMart)”
手机下单,快人一步
售后服务

扫描二维码
或添加“GeneGroup005”
获取更快速售后支持
PacBio MAS-Seq -- 开启肿瘤单细胞研究新时代

以下文章来源于PacBio研习社 ,作者PacBio

单细胞 RNA 测序(scRNA-seq)能够表征源自复杂组织的单个细胞之间的基因表达差异,提供比传统 RNA-seq 更高分辨率的信息。目前绝大部分的单细胞 RNA 测序都是基于 NGS 测序平台完成的,由于 NGS 测序短读长的原因,转录本均需要片段化后进行测序,只能得到转录本的各端的信息,得到的测序数据只能提供细胞内基因水平的表达信息,缺少可能对疾病或生物功能发挥十分关键作用的重要异构体多样性等信息。 为解决以上问题,PacBio 推出了 MAS-Seq 试剂盒,通过将短的 cDNA 进行串联建库的方式连接起来再进行HiFi测序,从而解决了长读长单细胞 RNA 测序的通量瓶颈: 完美适配 10x genomics 的 3’ 测序文库制备; 使用全长 cDNA 串联技术,将 HiFi reads 利用率提高了 16 倍; 结合 PacBio 特有的 Iso-Seq 测序,可以全面解析单细胞的可变剪切和融合基因等信息; 一个文库即可以获得 3000-10000 个细胞表达的 RNA 信息; 当搭配PacBio SMRTcell 8M 测序芯片时,可产生 4000 万条转录本序列; 当搭配最新的 SMRTcell 25M 测序芯片时,可产生 8000 万条转录本序列。 MAS-Seq 的发布,将使单细胞转录组研究从基因研究推进到转录本异构体的新领域,而近期,瑞士苏黎世联邦理工学院研究团队创新性地采用了与 MAS-Seq 类似的实验策略,测序得到了基于 PacBio 的全长 scRNA-seq 数据,与传统基于 NGS 的 scRNA-seq 测序相比,在测序成本差不多的情况下,获得了足够的单细胞全长转录本数据,通过分析发现 PacBio 全长单细胞转录组不仅可以对细胞类型进行准确的鉴定,同时还能发现二代单细胞转录组无法测到的融合基因表达,患者特异性和肿瘤组织特异性的 isoform 选用偏好性,以及能够发现更多外显子结合位点。这些信息的获取,为研究肿瘤的发生发展、克隆演化,提供了进一步探索的基础。 研究背景 肿瘤是一类复杂的疾病,基因组和转录组层面各维度的异常都可能参与到驱动肿瘤的形成和演变过程中,包括点突变、插入缺失、基因融合、基因拷贝数变异、可变剪接导致的不同转录异构体等等。除此之外,肿瘤内部的异质性及其与肿瘤微环境(TME)的相互作用,更是提升了其复杂程度。 基于 NGS 测序的 scRNA-seq,因为短读长的限制,一般只能观察细胞内基因的表达,为了进一步对肿瘤进行充分解析、逐个击破,基于 PacBio 单分子测序的单细胞全长转录组测序应运而生。不仅能够在单细胞水平揭示全基因组范围内细胞间的表达异质性,在临床更关注的突变、融合、拷贝数变异、可变剪接方面亦有优异表现。 材料方法 3个卵巢癌病人的肿瘤标本和癌旁配对样本 测序方案 1. 10× 单细胞捕获结合短读长测序,20,000-50,000 reads/cell。 2. PacBio 全长 scRNA-seq 测序,创新性的在流程中增加2轮biotin-PCR以减少数据中的模板转换寡核苷酸;将全长 cDNA 串联成 8-11kb 的长片段,大幅降低了测序成本,使 PacBio sequel II 的测序通量提高到 12,000 reads/cell。 3. 单细胞 DNA 测序,用于后续的融合基因验证。 主要结果 识别出大量的新的 isoform 基于 PacBio 的全长 scRNA-seq,共识别到 152,546 个转录异构体,其中 52,884 个为完整的新发现的转录异构体(下图NIC+NNC),其中 40,046 个被 GENCODE 数据库确认为有效,并且 42% 新发现的转录异构体具有蛋白编码功能。 研究人员还对细胞类型特异性和细胞特异性的isoform 进行了分析,发现在肿瘤细胞和间皮细胞中存在 isoform 亚型的差异表达。以基因 IGF1 (胰岛素样生长因子)为例(该基因与卵巢癌在内的多癌种的发展、进展、生存和耐药相关),来自所有患者的癌细胞几乎完全使用该基因的第二个外显子作为转录起始位点(II类亚型),而其他细胞主要使用第一个外显子(I类亚型),并且 IGF1 在癌细胞以及 TME 间皮细胞和成纤维细胞中表达明显高于远端间皮细胞和成纤维细胞。 2. 细胞类型准确鉴定 研究人员使用细胞特异的 marker 基因对 PacBio 全长 scRNA-seq 数据和基于 NGS 的 scRNA-seq 数据分别进行了细胞类型鉴定,发现两者单独鉴定的细胞类型和种类占比都非常相似。说明单独使用 PacBio 的全长 scRNA-seq 测序也能对细胞类型进行准确的鉴定。 3. 融合基因识别 通过 PacBio 的全长 scRNA-seq 数据能够更全面和准确地识别基因融合,在该研究中,发现在2号病例数据中有融合基因 IGF2BP2-TESPA1 高表达,该样本 PacBio 测序数据中有高达 2,174 条 reads 支持该结果,而在对应样本的短读长 scRNA-seq 数据分析中,该融合基因不仅没有被发现,还被错误的识别为 TESPA1 基因高表达,原因是短读长没有跨越和覆盖基因的融合位点,导致错误的分析结果。为确保分析结果的准确性,研究人员进一步通过 scDNA-seq 验证了这个融合基因的分析结果。 研究结论和展望

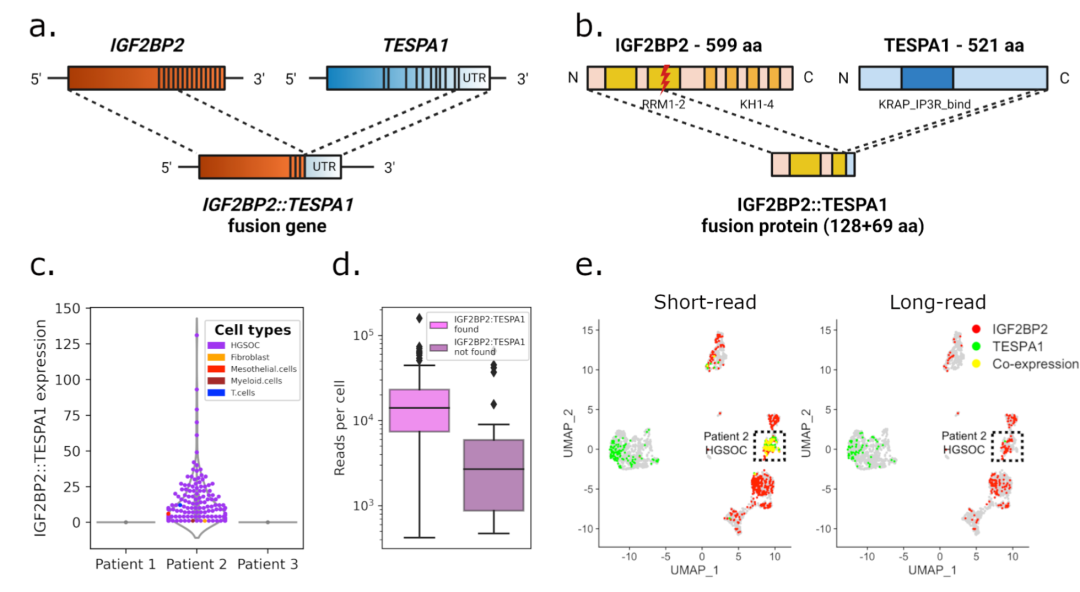

基于 PacBio 的全长 scRNA-seq 的临床应用将为癌症的形成、发生、TME形成、药物靶点和个体化的治疗提供比普通 scRNA-seq 更多的信息。之前由于 PacBio 的较高测序成本原因,全长 scRNA-seq 未被研究人员大范围使用,但是随着 PacBio 最新 Revio 的上市,测序成本已经显著降低,同时如果搭配 MAS-seq 试剂盒,将单条 HiFi reads 数据的利用率提高了 16 倍左右,使全长 scRNA-seq 的测序得到的有效 reads 进一步大幅提升,凭借该技术在检测突变、基因融合、可变剪接的覆盖范围和测序准确性方面的巨大优势,将为精准肿瘤学领域提供多种优势和新机会,也将为癌症疫苗的个性化药物预测和新抗原检测奠定坚实基础。
一 END 一
想了解更多有关 PacBio Mas-seq 是如何改变单细胞 RNA 测序的世界,请扫描下方二维码或直接访问:https://pacbio.cn/products-and-services/applications/rna-sequencing/single-cell-rna-sequencing/ 。

PacBio SMRT 测序只用于科研,不用于临床。











