
技术交流

扫描二维码
或添加“GeneGroup003”
获取更多更新资讯
商城订购

扫描二维码
或添加“基因商城(GeneMart)”
手机下单,快人一步
售后服务

扫描二维码
或添加“GeneGroup005”
获取更快速售后支持
FusionSeeker——长读长基因融合表征&融合转录本重建新工具来了!

融合基因分析新工具分享
结 果 模拟数据的基因融合检测 研究人员首先在模拟数据上对FusionSeeker基因融合检测的准确性进行了基准测试。随机生成150个具有不同的表达水平(10x、50x和100x)的基因融合转录本(100个在外显子中有断点,50个在内含子中有断点),分别用PacBio Iso-Seq和Nanopore测序技术进行测序,得到的reads均用3款长读长基因融合caller——FusionSeeker、JAFFAL和LongGF进行基因融合检测分析。 在三个基因融合caller中,FusionSeeker识别出的基因融合事件比其他两个caller更多,始终具有最高的F1分数(表1)。基于Iso-Seq数据进行FusionSeeker分析的F1分数更高,主要得益于其检测内含子区域基因融合的能力。其中FusionSeeker识别了94.67%的内含子事件,而JAFFAL和LongGF分别只识别了14.67%和54.67%(表1-1和表1-2)。 表1-1 三种基因融合caller分析Iso-seq和Nanopore数据中基因融合事件的准确性 表中的召回率、精度和F1得分为模拟数据集三次重复的平均值。三个基因融合caller中召回率、精度和F1分数最高的标记为粗体。 表1-2 三种基因融合caller报告的Iso-seq数据中内含子/外显子中的基因融合事件
为了生成高精度的转录本序列,FusionSeeker使用包含融合基因的reads进行偏序对齐,生成每个基因融合事件的一致性序列,并评估基因融合序列重建的准确性。 在模拟数据中,FusionSeeker重构了超过99.5%融合事件的全长转录本,使用Iso-Seq和Nanopore reads的平均序列识别率分别为99.87%和99.14%(表2)。当与参考基因组对齐时,FusionSeeker转录本序列显示出比原始reads更好的一致性(图1)。相比与原始序列,PacBio Iso-Seq数据(左)通过偏序比对生成的融合基因的序列比Nanopore数据(右)显示了更高的一致性(图1)。综上所述,FusionSeeker可以准确地识别基因融合并重建全长融合转录本序列。 表2 模拟数据上FusionSeeker重建的融合转录本事件 当95%以上的模拟融合转录本序列被重建时,将FusionSeeker输出的转录本序列视为“全长”。Detected GF,每个数据集中检测到的基因融合数。Full-length transcript,与全长转录本序列融合的基因数量。Identity,重建的转录序列与真实序列的一致性。 图1 原始序列和FusionSeeker重建的融合基因序列的一致性分析。
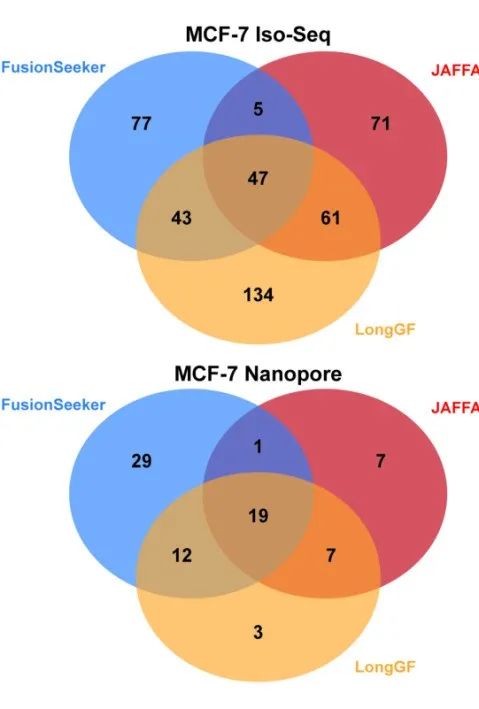
癌症转录组中的基因融合发现 然后,研究人员将这三种基因融合caller应用于MCF7癌细胞系。将MCF7癌细胞系的PacBio Iso-Seq和Nanopore reads都与人类参考基因组进行比对。 在MCF-7细胞系中,FusionSeeker在Iso-Seq数据集中识别出172个基因融合,在Nanopore数据集中识别出61个基因融合,其中分别有21个和20个基因融合事件已被验证(表3)。当比较三个caller的基因融合检测能力时,在Iso-Seq和Nanopore数据中,三个caller分别报告了47和19个基因融合(图2B)。 综上,相比于Nanopore测序技术,基于Iso-Seq测序能够更加均匀地覆盖转录本全长,允许检出更多的基因融合事件。 表3. 癌症细胞系中先前验证的基因融合的检测 图2. 癌症细胞系的基因融合发现。 为了证明其临床实用性,研究人员还在一个急性髓系白血病(AML)患者样本上应用了FusionSeeker。FusionSeeker识别出RUNX1和RUNX1T1之间的一个预验证的基因融合,并报告了该患者样本中另外7个可信的基因融合事件(表4)。 表4 在AML患者样本中检测到基因融合 重建融合基因序列的评估 最后研究人员评估了由FusionSeeker生成的转录本序列。在MCF-7细胞系的Iso-Seq和Nanopore数据中,FusionSeeker转录本序列与参考基因序列的一致性显著提高,其中基于Iso-Seq生成的转录本序列更接近原始序列(图3)。 图3. 比较MCF-7细胞系的Iso-Seq和Nanopore数据中FusionSeeker重建的融合转录本序列与参考基因序列的一致性。 值得注意的是,通过与参考序列比较得出的一致性是对转录本序列准确性的低估,这是由于这些细胞系中存在遗传变异。这些遗传变异通常会在转录本序列中保留(图3)。在Iso-Seq和Nanopore数据集中,C/T的SNP信息均保留在chr14:37592537区域的基因融合重建转录序列中。但是在这两个测序数据中,与Nanopore数据(图4B)相比,Iso-seq数据(图4A)的测序错误和gap更少(图4)。 图4. MCF-7细胞系Iso-Seq (A)和nanopore(B)数据中基因融合事件FOXA1:TTC6的IGV视图。





讨 论 因此,FusionSeeker是用于长读长癌症转录组测序数据中外显子和内含子区域基因融合全面检测的更优方法,PacBio Iso-seq长读长测序则是用于转录本准确测序的更优技术。结合FusionSeeker和PacBio Iso-seq有助于发现更多新的基因融合事件,这些新的基因融合可能会对肿瘤的发生和进展有重要意义,促进下游功能和临床研究。
HiFi测序新款机型——Revio于10月25日正式上市,保持HiFi测序的高准确度和长读长的基础上:测序通量在sequel IIe的基础上提升15倍;一次运行可即可获得360 Gb HiFi数据;搭载NVIDIA GPUs和Google DeepConsensus,计算能力与Sequel IIe相比提升20×;测序时间由原来的30小时降低到24小时。更好助力您的研究!!! 通量更高成本更低的Revio您不来了解一下吗?










