技术交流

扫描二维码
或添加“GeneGroup003”
获取更多更新资讯
商城订购

扫描二维码
或添加“基因商城(GeneMart)”
手机下单,快人一步
售后服务

扫描二维码
或添加“GeneGroup005”
获取更快速售后支持
PacBio HiFi Reads---极大提升病毒宏基因组研究中的样本覆盖度和多样性

PacBio三代测序技术,其HiFi Reads以长读长(最长可达25 kb)、高准确率(≥Q30,即超过99.9%的测序精度)、单分子分辨率、高灵敏度、无GC偏好性等优势,无论是在医学领域、动植物基因组还是在微生物研究领域中,都在崭露头角,成为基因组学研究技术的新宠。 今天,就让我们通过“Long-Read Metagenomics Improves the Recovery of Viral Diversity from Complex Natural Marine Samples” 一文,一起看看PacBio HiFi测序技术在提高复杂天然海洋样品中病毒多样性和覆盖度研究中的优势及应用。 近日,西班牙团队的科学家通过使用PacBio HiFi reads进行病毒宏基因组测序,探索了环境病毒的巨大遗传多样性。本研究的目的是比较长读长测序与使用 Illumina (短读长)经典方法用于病毒群落的研究效率,结果发现PacBio测序技术可以大大提高病毒群体代表性样本的覆盖度,避免了数据组装中的偏差,也使基于病毒标记蛋白的病毒基因多样性有了更好的发现。 + + + + + + + + + + + 研究背景 海洋病毒是海洋环境中最丰富的生物实体,估计其种群密度为每毫升海水107个,因此它们是海洋中驱动生物地球化学循环的关键因素,它们既通过捕食浮游植物和异养细菌的副产物释放有机物,也可通过病毒感染操纵宿主代谢。 在过去的20年中,由于短读长(SR)测序的进步,宏基因组学已经推动了海洋环境中病毒群落的研究。而PacBio的HiFi长读长(LR)测序技术的出现既解决了困扰SR测序研究多年的高多样性微生物的低回收率的问题,可实现非常高的覆盖率,同时还具有较低的错误率,为原核生物群落在其自然环境中的研究开辟了新的途径。 + + + + + ✦ + + 结果及数据表现
✦+
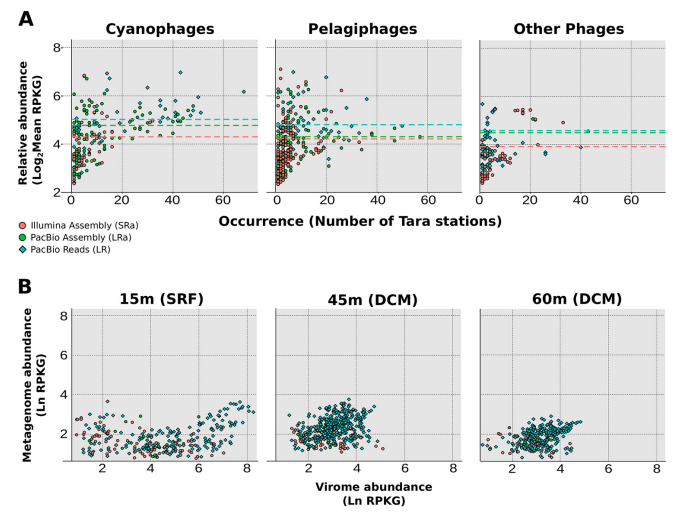
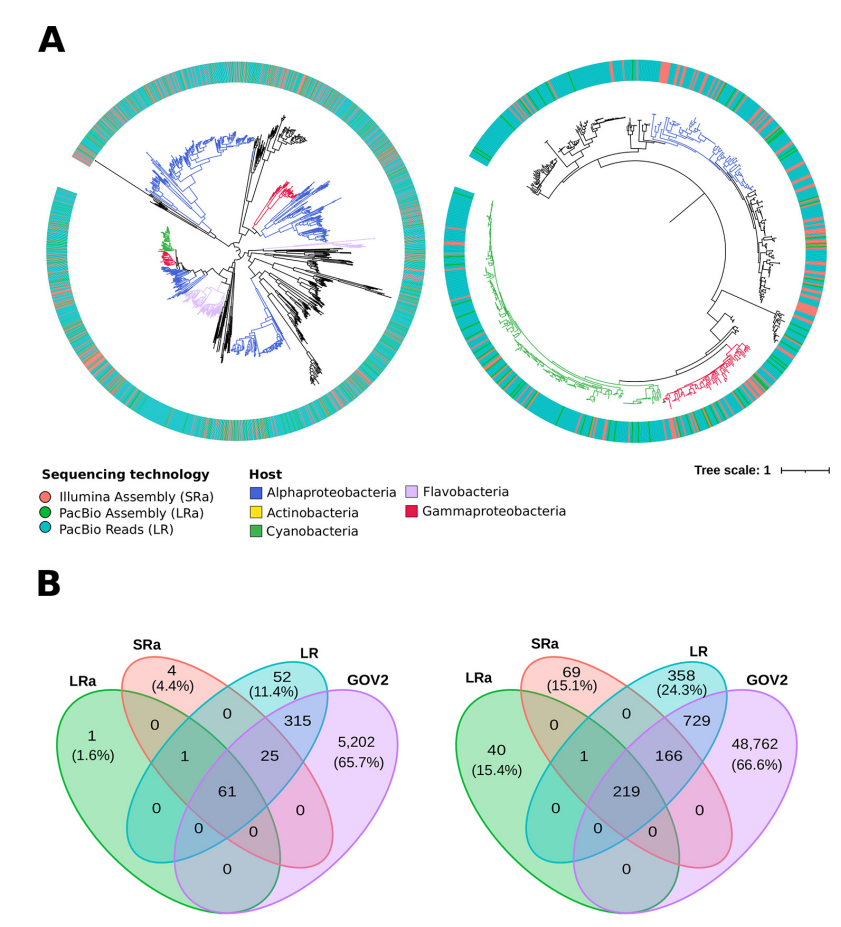
+ 1、病毒序列恢复与统计 为了评估LR对宏基因组学的病毒基因组多样性的分辨能力,并将其与SR测序进行比较,作者分析了来自冬季地中海近海水域的单个海洋样本。处于分裂周期细胞内的病毒复制产生了一种自然扩增,这使得在宏基因组样品中发现大量病毒起源序列成为可能。结果如下图所示,该样品用Illumina和PacBio Sequel II系统测序,然后组装两次,首先仅使用 Illumina 短读长数据组装成短读长组装数据集(SRa) ,然后使用Illumina和PacBio长读长混合组装,产生长读长数据集(LRa)。同时为了评估数据组装过程可能引入的偏差,作者还分析了单独使用PacBio CCS(HiFi) reads组装的数据结果(LR)。 首先通过对所有序列运行VIBRANT来识别每个数据集中的病毒来源,总共恢复了54,082个推定的病毒序列(SRa 中的10,979个,LRa中的947个和LR中的42,156个),如上表所示。为了评估不同的组装方法是否恢复了相同的病毒群落,作者通过比较三个数据集彼此来确定每个数据集中的独特序列。大多数来自LRA的序列也在SRa中发现,但值得注意的是,尽管SRa数据集包含相当数量的独特序列(5,886) ,但绝大多数独特序列在LR数据集中被发现(30,203; 总病毒 LR测序的71%) ,这一组装结果揭示了PacBio HiFi测序技术极大的提升了病毒基因组的多样性,发现了大量独特的病毒基因组序列,这是通过之前的技术手段无法发现的。当编码末端酶大亚基(terL)的标记基因的结果与包含393个独特末端酶基因(以95%氨基酸同一性聚类)的LR数据集相比较时,也存在这种多样性差异,而SRa和LRa数据集分别为30和2。 接下来,作者感兴趣的是评估这种新的多样性是否已经被以前的研究捕获,所以将三个数据集与迄今为止最大的海水噬菌体数据库——全球海洋病毒2(GOV2)数据集进行比对分析,发现整个数据集(SRa,LR和LRa)中有30,997个在GOV2中未发现的病毒序列,且其中绝大多数(26,766)来自于LR数据集。 + 2、PacBio HiFi测序策略 提高海洋样品的检测丰富度 接下来,作者为了分析只在LR data set 中出现的多样性数据是否是丰富的且是在自然界中存在的,参考对比整个塔拉海洋(Tara Oceans)宏基因组数据集,进行了 SRa,LRa和LR病毒序列的募集分析。如果一段sequence有5条Kb以上的reads数(RPKG:每千兆级reads中来自于某基因每千碱基长度的reads数),则我们考虑这段序列存在于宏基因组样品中,且确认度为95% ,contig覆盖率为50% 。如下图(A)所示,病毒序列的相对丰度通过它们在来自塔拉海洋的噬藻体(cyanophages),深海噬菌体(pelagiphages)和其他噬菌体的宏基因组数据的招募值来测量。X轴显示了累积超过覆盖阈值的 Tara stations 的数量,而y轴显示了组合招募值( RPKG 中)。圆圈表示不同数据组装方式(绿色表示混合组装,橙色表示Illumina组装) ,蓝色表示原始PacBio reads。每个数据集的募集算法用直线表示,下图中显示在所有三种情况下(pelagiphages, Cyanobacteria, and other phages) , LR覆盖到的序列比其它组装结果的丰富度更高。 + 3、LR数据中发现更高的 病毒宏基因组多样性 由于发现了LR中有大量的病毒序列不包含在其他数据集中(可能在组装过程中丢失) ,并且在自然界中非常丰富,作者决定进一步分析这种多样性。鉴于没有用于分析病毒多样性的通用标志物,本文使用了许多不同的噬菌体特异性标志物(末端酶大亚基[ terL ] 、复制性 DNA 解旋酶[ dnaB ] 、尾管蛋白,主要衣壳蛋白和噬菌体裂解蛋白spanin)以及几个表征良好的辅助代谢基因(AMG)(胸苷酸合成酶[[thyX] 、磷酸化肝素异构酶[ gmhA ] 、核糖核苷二磷酸还原酶[ nrdA ] 、核糖核苷酸还原酶大亚基和磷饥饿诱导蛋白[ phoH ])。 通过构建系统发育树以及与GOV2数据库的比对,分析了以上所选markers在同一样本中的三个数据集中的多样性(上图所示) 。系统发育树表明,所有的噬菌体进化分支都不仅仅由LR获得的独特蛋白质组成,因此可以得出结论,从LR数据集中覆盖到的独特序列不属于新的噬菌体分类群,而是属于已知的进化分支。比较三个数据集之间的独特蛋白质分布,LR数据集通常包含比其它组装数据集更多的独特序列。此外,独特变异的百分比总是在LR中较高。需要强调的是,LR没有揭示新的噬菌体分支的事实并不意味着与它们的新颖性不相关(比如关于内溶素功能研究的案例),这种尚未开发的多样性可能会助力于生物技术,例如寻找用于医药的生物制剂,以及将生物工业应用于农业或食品生产。 参考文献: Long-Read Metagenomics Improves the Recovery of ViralDiversity from Complex Natural Marine Samples.Asier Zaragoza-Solas et al.May/June 2022 Volume 7 Issue 3. 点击链接,了解更多 // 基因有限公司作为PacBio公司在中国区的独家代理商,自2011年以来将PacBio第三代单分子实时测序技术引入国内,一直为国内用户提供专业的三代测序系统的安装培训,技术支持,应用培训与售后维护工作,赢得客户的一致好评与信任。基因有限公司将一如既往的支持越来越多的PacBio用户。